 Datanami
Datanami EnterpriseAI
EnterpriseAI HPCwire Japan
HPCwire Japan QCwire
QCwire HPC & AI Wall Street
HPC & AI Wall StreetAdvances in sequencing technology have significantly increased data generation and require commensurate computational advances for bioinformatics analysis. Advanced architectures based on reconfigurable computing can reduce application run times from hours to minutes and address problem sizes unattainable with commodity servers. The increased capability also fundamentally improves research quality by allowing more accurate, previously impractical approaches. The use of a hybrid-core computing architecture can be used to solve data-intensive problems of next-generation sequencing analysis like de novo assembly and reference mapping of short-read sequences.
Two important steps in next-generation sequencing analysis are de novo assembly and reference mapping of short-read sequences. Both of these lend themselves to high levels of acceleration with the FPGA-based coprocessor on the Convey systems. Convey’s bioinformatics applications Graph Constructor and BWA can be used in conjunction with, or replace, workflows using standard Velvet[1] and BWA[2], respectively. Graph Constructor reduces not only run time for Velvet, but also reduces memory requirements, making it capable of larger assemblies. Additional performance and workflow optimization includes a fast kmer counting tool that allows quick identification of optimal kmer length and coverage cutoffs for de novo assembly.
Convey’s Hybrid Core Architecture: Fast Compute, Faster Memory
The Convey Hybrid-Core (HC) architecture pairs Intel® x86 microprocessors with a coprocessor comprised of reconfigurable hardware (FPGAs) (Figure 1). Algorithms are implemented as instructions, called personalities, which are loaded onto the FPGAs at runtime to accelerate the applications that use them. Complementing the high performance of the reconfigurable compute elements, Convey’s hybrid-core system also has a highly parallel memory subsystem that is optimized for random accesses. Hybrid-Core Globally Shared Memory (HCGSM) provides a single coherent view of memory to the cache based x86 cores and the high throughput word optimized processing elements on the coprocessor. Bioinformatics applications that experience memory performance limitations on cache-based x86 servers greatly benefit from Convey’s memory architecture.
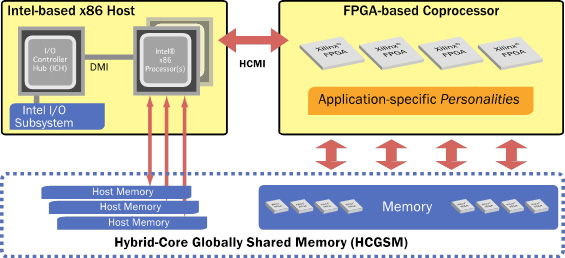
Figure 1. The Convey Hybrid-Core Architecture. The architecture pairs an Intel x86 host system tightly integrated with a reconfigurable FPGA based coprocessor. Hybrid-Core Globally Shared Memory (HCGSM) provides a single coherent view of memory to the x86 cores and the coprocessor’s highly parallel memory subsystem.
Burrows-Wheeler and de Bruijn Graph Personalities
de Bruijn graph-based assemblers such as Velvet consist of large numbers of relatively simple operations on large randomly accessed data structures. Conventional architectures lack sufficient parallelism in the core processing elements and the memory subsystem to efficiently execute these algorithms. The Convey Graph Constructor implements a high speed de Bruijn graph generator that can reduce the runtime and memory footprint for graph-based genome assembly. It can be run by itself or in conjunction with the Velvet application.
Other algorithms also benefit from massively parallel implementations of application-appropriate-data-type operations, which use logic gates more efficiently than commodity servers. In Burrows-Wheeler mapping applications, significant gains are made in the population bit count required to traverse the compressed reference suffix trees in memory. Convey has developed a personality that improves the performance of the aln step of the BWA processing pipeline, and a version of the open-source BWA application with thread parallelized single- and paired-end processing. The BWA personality has 64 alignment units which each operate on 32 sequences simultaneously, for a total of 2,048 simultaneous alignment operations.
Align and Paired End Performance for Human Genome
For these tests (Figure 1) we aligned paired-end sequence data from the 1000 Genomes project to a human reference (human_g1k_v37), consisting of 84 sequences and a total of 3.1 billion bases. SRR189815_1 and SRR189815_2 are paired-end Illumina reads from individual HG00124 containing a total of 242 million reads, average length 101. The aln steps were run using Convey accelerated BWA on HC-1 and HC-1ex systems, and the paired end step was run on a commodity x86 system using a parallelized version of bwa sampe. The results are compared to BWA 0.5.9 running on the commodity server.
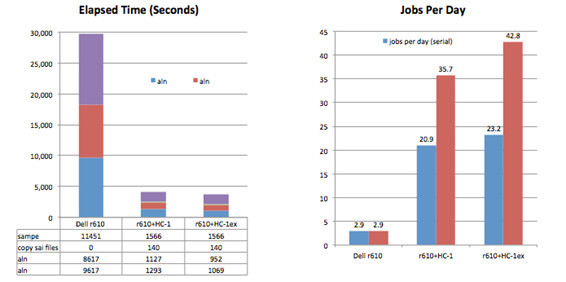
Figure 1. Align and Paired End Performance for Human Genome. The addition of an HC-1ex and the Convey accelerated BWA pipeline to a commodity x86 system delivers 14.7x the throughput of the x86 system alone, processing 120 K reads/sec.
Results
- Convey’s hardware accelerated aln is 7.5x (HC-1) and 9x (HC-1ex) over a 12-core x86.
- Thread parallel sampe is 7.3x faster than the standard bwa implementation on the same hardware.
- The addition of an HC-1ex and the Convey accelerated BWA pipeline to a commodity x86 system delivers 14.7x the throughput of the x86 system alone, processing 120 K reads/sec.
de novo Assembly Parameter Optimization
A feature of the Convey Bioinformatics Suite is the Kmer Counter. The Convey Kmer Counter generates a histogram of kmer coverage counts by hashing kmers in each read sequence. As shown in Figure 2, analysis of the read data aids in selecting optimal kmer length and coverage cutoff values for de novo assembly.
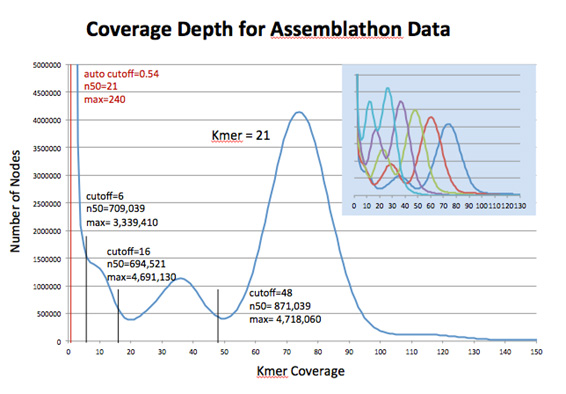
Figure 2. Histogram of kmer coverage for the Assemblathon data set, as produced by Convey’s Kmer Counter for kmer length 21. Statistics for assembly results using the selected coverage cutoffs show the impact of parameter selection on assembly quality, as compared with Velvet’s default setting. Convey’s Kmer Counter can analyze multiple kmer lengths in the same run as shown in the blue overlay.
Results:
- Higher quality assemblies by using optimal parameters
- Reduced run time and memory by avoiding poor kmers
- Extremely efficient compared with VelvetOptimiser
- Handles longer kmer lengths than Jellyfish
- Analyzes multiple kmer lengths in a single job
Summary
Convey has developed a personality that improves the performance of the aln step of the BWA processing pipeline, and a parallelized version of the samse and sampe processing steps, that allow Convey systems to dramatically reduce time to solution and increase throughput 15x for a full BWA paired-end pipeline, processing 120 K reads/sec.
We have developed a GraphConstructor personality that interfaces to Velvet and Oases that reduces memory requirements by about 75% and accelerates throughput by an order of magnitude, making it possible to tackle previously impractical genomes with higher quality results. In addition to this work, there are several other projects recently submitted or in progress comparing the performance and accuracy of Convey’s Graph Constructor for genome and transcriptome assemblies, comparing with a range of popular assembly programs.
We are working on additional performance and workflow optimization for these applications, as well as accelerating additional applications.
References and Acknowledgements
- “Velvet: Algorithms for de novo Short Read Assembly Using de Bruijn Graphs”, Daniel R. Zerbino and Ewan Birney, EMBL-European Bioinformatics Institute, Genome Res. 18 (2008) 821.
- “Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform”, Heng Li and Richard Durban, Wellcome Trust Sanger Institute, Bioinformatics 25 (2009) 1754.
- “Metagenomic discovery of biomass-degrading genes and genomes from cow rumen”, Hess ,et al, Science 331 (2011) 463.
- “Efficient Graph Based Assembly of Short-Read Sequences on a Hybrid Core Architecture“ Alex Sczyrba, Abhishek Pratap, Shane Canon, James Han, Alex Copeland, Zhong Wang, DOE Joint Genome Institute User Meeting, March 2011.
For more information go to Convey Computer.


























































