 Datanami
Datanami EnterpriseAI
EnterpriseAI HPCwire Japan
HPCwire Japan QCwire
QCwire HPC & AI Wall Street
HPC & AI Wall Street
Cancer, the second-leading cause of death in the U.S. after heart disease, kills more than 500,000 citizens per year, including about 2,000 children.
In 2016, then Vice President Joe Biden launched the Cancer Moonshot, saying: “I know that we can help solidify a genuine global commitment to end cancer as we know it today — and inspire a new generation of scientists to pursue new discoveries and the bounds of human endeavor.”
The importance of high performance computing (HPC) in cancer research was recognized by the Cancer Moonshot Task Force report, and by then Vice President Joe Biden and Energy Secretary Ernie Monitz.
“Supercomputers are key to the Cancer Moonshot,” Monitz wrote. “These exceptionally high-powered machines have the potential to greatly accelerate the development of cancer therapies by finding patterns in massive datasets too large for human analysis. Supercomputers can help us better understand the complexity of cancer development, identify novel and effective treatments, and help elucidate patterns in vast and complex data sets that advance our understanding of cancer.”
With complex, non-linear signaling networks, multiscale dynamics from the quantum to the macro level, and giant, complex datasets of patient responses, cancer is quite possibly the ultimate in HPC problems.
“What could be more complicated and more important?” said J. Tinsley Oden, a computational researcher at The University of Texas at Austin applying uncertainty quantification to cancer treatment predictions. “At each step, it has the most complex features. It is really a garden of rich, important problems that are in the path of many of the developments that we’ve been working on for years.”

Hundreds of oncologists, biologists and computer scientists use the HPC systems at the Texas Advanced Computing Center (TACC) to understand the fundamental nature of cancer biology and to improve cancer treatments. Their work addresses a range of cancers types and treatment modalities, and spans applied or fundamental research.
Though diverse in their specific targets, the approaches they use can be loosely grouped into seven broad methodologies: molecular simulation; bioinformatics; mathematical modeling; computational treatment planning; quantum calculation; clinical trial design; and machine learning. The following sections describe and provide examples of each.
Molecular Simulations
Simulating protein and drug interactions at the molecular level enables scientists to understand the mechanics of cancer to design more effective treatments.
For Rommie Amaro, professor of Chemistry and Biochemistry at the University of California, San Diego, this means uncovering new pockets in tumor protein 53 (p53) — “the guardian of the genome” — which plays a crucial role in conserving the stability of DNA and preventing mutations.
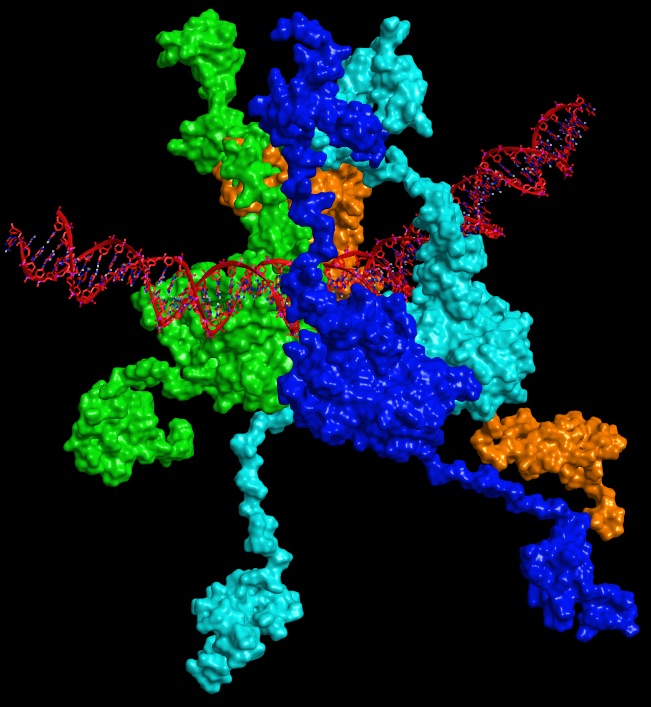
In September 2016, writing in the journal Oncogene, Amaro reported results of the largest atomic-level simulation of the p53 to date — comprising more than 1.5 million atoms. The simulations, enabled by the Stampede supercomputer at TACC, helped identify new binding sites on the surface of the protein that could potentially reactivate p53.
“When most people think about cancer research they probably don’t think about computers,” she said. “But biophysical models are getting to the point where they have a great impact on the science.”
Virtual drug screening is another important HPC application for cancer research. Shuxing Zhang, professor of experimental therapeutics at MD Anderson Cancer Center, used molecule simulations on TACC’s Lonestar5 system to screen 1,448 Food and Drug Administration-approved small molecule drugs to determine which had the molecular features needed to bind and inhibit TNIK — an enzyme that plays a key role in cell signaling in colon cancer.
Zhang discovered that mebendazole, an FDA-approved drug that fights parasites, could effectively bind to TNIK and inhibit its enzymatic activity. He reported his results in Nature Scientific Reports in September 2016.
“Such advantages render the possibility of quickly translating the discovery into a clinical setting for cancer treatment in the near future,” Zhang wrote.
Bioinformatics
The human genome consists of three billion base pairs, so identifying single mutations by sight simply isn’t possible. For that reason, the field of bioinformatics — which uses computing and software to identify patterns and differences in biological data — has been an enormous boon for cancer researchers.
But bioinformatics is more than simple, one-to-one pattern matching.
For Vishy Iyer, a molecular biologist at The University of Texas at Austin (UT Austin), and his collaborators, access to TACC’s Stampede supercomputer helps them mine reams of data from The Cancer Genome Atlas to identify genetic variants and subtle correlations that affect gene expression in tumors.
“TACC has been vital to our analysis of cancer genomics data, both for providing the necessary computational power and the security needed for handling sensitive patient genomic datasets,” Iyer said.
In February 2016, Iyer and a team of researchers from UT Austin and MD Anderson Cancer Center reported in Nature Communications on a genome-wide transcriptome analysis of the two types of cells that make up the prostate gland. They identified cell-type-specific gene signatures that were associated with aggressive subtypes of prostate cancer and adverse clinical responses.
“This knowledge can be helpful in the development of more targeted therapies that seek to eliminate cancer at its origin,” Iyer said.
Using a similar methodology, Iyer and a team of researchers from UT Austin and the National Cancer Institute identified a transcription factor associated with an aggressive type of lymphoma that is highly correlated with poor therapeutic outcomes. They published their results in the Proceedings of the National Academy of Sciences in January 2016.
Whereas Iyer, an experienced HPC user, develops custom tools for his analyses, a much larger number of researchers access Stampede and comparable systems through scientific gateways. One prominent gateway is Galaxy, an open source bioinformatics platform that serves 30,000 researchers and runs more than 3,000 compute jobs a day.
Since 2014, TACC has powered the data analyses for a large percentage of Galaxy users, allowing researchers to solve tough problems in cases where their personal computer or campus cluster is not sufficient. Of those researchers, a significant subset use the site to analyze cancer genomes.
“Galaxy can be used to identify tumor mutations that drive cancer growth, find proteins that are overexpressed in a tumor, as well as for chemo-informatics and drug discovery,” said Jeremy Goecks, Assistant Professor of Biomedical Engineering and Computational Biology at Oregon Health and Science University and one of Galaxy’s principal investigators.
Goecks estimates that hundreds of researchers each year use the platform for cancer research, himself included. Because cancer patient data is closely protected, the bulk of this usage involves either publically available cancer data, or data on cancer cell lines – immortalized cells that reproduce in the lab and are used to study how cancer reacts to different drugs or conditions.
“This is an ideal marriage of TACC having tremendous computing power with scalable architecture and Galaxy coming along and saying, we’re going to go the last mile and make sure that people who can’t normally use this hardware are able to.”
Mathematical Modeling
While some researchers believe bioinformatics will rapidly advance the understanding and treatment of cancer, others think a better approach is to mathematize cancer: to uncover the fundamental formulas that represent how cancer, in its varied forms, behaves.
At the Center for Computational Oncology at UT Austin, researchers are developing complex computer models to predict how cancer will progress in a specific individual.
Each factor involved in the tumor response — whether it is the speed with which chemotherapeutic drugs reach the tissue or the degree to which cells signal each other to grow — is characterized by a mathematical equation that captures its essence. These models are combined and parameterized and initialized with patient-specific data.
In April 2017, writing in the Journal of The Royal Society Interface, Thomas Yankeelov and collaborators at UT Austin and Vanderbilt University, showed that they can predict how brain tumors (gliomas) will grow in mice with greater accuracy than previous models by including factors like the mechanical forces acting on the cells and the tumor’s cellular heterogeneity.
To develop and implement their mathematically complex models, the center’s scientists use TACC’s supercomputers, which enable them to solve bigger problems that they otherwise could and reach solutions far faster.
Recently, the group has begun a clinical study to predict, after one treatment, how an individual’s cancer will progress, and use those predictions to plan the future course of treatment.
“There are not enough resources or patients to sort this problem out because there are too many variables. It would take until the end of time,” Yankeelov said. “But if you have a model that can recapitulate how tumors grow and respond to therapy, then it becomes a classic engineering optimization problem. ‘I have this much drug and this much time. What’s the best way to give it to minimize the number of tumor cells for the longest amount of time?’”
Computing at TACC helps Yankeelov accelerate his research. “We can solve problems in a few minutes that would take us three weeks to do using the resources at our old institution,” he said. “It’s phenomenal.”
Quantum Calculations
X-ray radiation is the most frequently used form of radiation therapy, but a new treatment is emerging that uses a beam of protons to kill cancer cells with minimum damage on surrounding tissues.
“As happens in cancer therapy, we know empirically that it works, but we don’t know why,” said Jorge A. Morales, a professor of chemistry at Texas Tech University and a leading proponent of the computational analysis of proton therapy. “To do experiments with human subjects is dangerous, so the best way is through computer simulation.”
Computational experiments can mimic the dynamics of the proton-cell interactions without causing damage to a patient and can reveal what happens when the proton beam and cells collide from start to finish, with atomic-level accuracy. Morales has been simulating proton-cell chemical reactions using quantum dynamics models on TACC’s Stampede supercomputer to investigate the fundamentals of the process.
His studies, reported in PLOS One in March 2017, as well as in Molecular Physics, and Chemical Physics Letters (2015 and 2014 respectively), have determined the basic byproducts of protons colliding with water within the cell, and with nucleotides and clusters of DNA bases – the basic units of DNA. The studies shed light on how the protons and their water radiolysis products damage DNA.
Though fundamental in nature, the insights and data that Morales’ simulations produce help researchers understand proton cancer therapy at the quantum level, and help modulate factors like dosage and beam direction.
“These simulations will bring about a unique way to understand and control proton cancer therapy that, at a very low cost, will help to drastically improve the treatment of cancer patients without risking human subjects,” Morales said.
Computational Treatment Planning
Wei Liu, a researcher at the Mayo Clinic, also studies proton therapy, but he looks at the treatment from a clinical perspective.
In comparison with current radiation procedures, proton therapy saves healthy tissue in front of and behind the tumor. It is particularly effective when irradiating tumors near sensitive organs where stray beams can be particularly damaging.
However, the pinpoint accuracy required by the protein beam, which is its greatest advantage, means that it must be precisely calibrated and that discrepancies from the ideal (whether from device, human error or even patient breathing) must be taken into consideration.
Writing in Medical Physics in January 2017, Liu and his collaborators showed that their “chance-constrained model” was better at sparing organs at risk than current methods.
“Each time, we try to mathematically generate a good plan,” he said. “There are 25,000 variables or more, so generating a plan that is robust to these mistakes and can still get the proper dose distribution to the tumor is a large-scale optimization problem.”
The researchers used the Lonestar5 supercomputer at TACC to generate treatment plans that minimize the risk and uncertainties involved in proton beam therapy.
“It’s very computationally expensive to generate a plan in a reasonable timeframe,” he continued. “Without a supercomputer, we can do nothing.”
Computational Trial Design
Another way researchers use TACC’s advanced computers is to design clinical trials that can better determine which combination of dosages will be most effective, specifically for the biological agents used in immunotherapy, which work very differently from chemotherapy and radiation.
Writing in the Journal of the Royal Statistics Society Series C (Applied Statistics), Chunyan Cai, assistant professor of biostatistics at McGovern Medical School at The University of Texas Health Science Center at Houston (UTHealth) described her efforts using Lonestar5 to identify biologically optimal dose combinations for agents that target the PI3K/AKT/mTOR signaling pathway, which has been associated with several genetic aberrations related to the promotion of cancer.

They investigated six different dose-toxicity and dose-efficacy scenarios and carried out 2,000 simulated trials for each of the designs.
Based on those simulations, she concluded that “the design proposed has desirable operating characteristics in identifying the biologically optimal dose combination under various patterns of dose–toxicity and dose–efficacy relationships.”
The research is leading to new, safer and more effective ways to test combinations of immunotherapeutic agents.
Machine Learning
A final, and truly radical, way that researchers are using HPC for cancer research is through the application of machine and deep learning.
The Eberlin research group at UT Austin develops clinical applications of ambient mass spectrometry for cancer diagnosis. They create tools and techniques to assist surgeons in distinguishing between normal and cancer tissue during tumor resection operations.
To do so, they have had to develop statistical methods that can analyze and interpret large amount of mass spectrometry data gathered from clinical samples.
Jonathan Young, a post-doctoral research in the group, is building machine learning classifiers to reliably predict whether a given tissue sample is cancer or normal, and if it is indeed cancer, which specific subtype the tumor belongs to.
Young uses the Maverick system at TACC, which contains a large number of NVIDIA GPUs, to develop and implement the machine learning algorithms. “The large memory capacity of Maverick is well suited for our extensive datasets, and the parallelization capability will aid in parameter sweeps during the training of classifiers,” Young said.
Young will present his work at the American Society for Mass Spectrometry (ASMS) Annual Conference this June.
Another example of the application of machine learning to cancer can be found in the work of Daniel Lobo, an assistant professor of biology and computer science at the University of Maryland, Baltimore County (UMBC). He is using machine learning to map out the cellular communication networks that underlie cancer, and to design methods to disrupt them.
In their January 2017 paper in Scientific Reports, Lobo and collaborators showed that machine learning can uncover the cellular networks that determine pigmentation in tadpoles and reverse-engineering never-before-seen coloration. Their work was facilitated by Stampede, which enabled the team to run billions of simulations to identify models of the cellular network and the means of altering it.
Lobo’s lab is applying the method to cancer research to determine what type of interventions might stop metastasis in its tracks without damaging other cells.
“Traditional approaches like chemotherapy attack the cells that grow the most, but leave cells that are signaling others to grow and that may be the most important,” Lobo says. “We’re using machine learning to find out the communication networks between these cells and hopefully to discover a treatment that can cause the tumor to collapse.”
“Getting a true understanding, given the complexity of the information, without some assistance from machine learning, is probably hopeless,” said Michael Levin, Lobo’s collaborator. “I think it’s inevitable that we use machine learning to enrich scientific and biomedical discovery.”
From patient-specific treatments to immunology to drug discovery, advanced computing is accelerating the basic and applied science underlying our understanding of cancer and the development and application of cancer treatments.
If scientists are the rocket in the cancer moonshot, HPC processing power is the jet fuel.
About the Author
Aaron Dubrow joined TACC in October 2007 as the Science and Technology Writer with the responsibility of reporting on the myriad of research and development projects undertaken by TACC.

























































