 Datanami
Datanami EnterpriseAI
EnterpriseAI HPCwire Japan
HPCwire Japan QCwire
QCwire HPC & AI Wall Street
HPC & AI Wall Street
IBM yesterday reported in Nature Communications the use of a novel algorithm to simulate BeH2 (beryllium-hydride) on a quantum computer. This is the largest molecule so far simulated on a quantum computer. The technique, which used six qubits of a seven-qubit system, is an important step forward and may suggest an approach to simulating ever larger molecules.
“Instead of forcing previously known classical computing methods onto quantum hardware, the scientists reversed the approach by building an algorithm suited to the capability of the current available quantum devices. This allows for extracting the maximal quantum computational power to solve problems that grow exponentially more difficult for classical computers,” according to the IBM announcement.
Quantum chemistry has long been regarded as of the great promises of quantum computing. A good example is nitrogen fixation – essentially making ammonia – from the air. Bacteria do it effortlessly. Industry still does it with a hundred-year-old Haber process, which is used mostly in fertilizer production today.
 Today, simulating even small molecules with the needed accuracy to predict energy states and reactivity is hard. IBM performed the numerical simulation on H2, LiH, and BeH2. “While this model of BeH2 can be simulated on a classical computer, IBM’s approach has the potential to scale towards investigating larger molecules that would traditionally be seen to be beyond the scope of classical computational methods, as more powerful quantum systems get built,” noted IBM.
Today, simulating even small molecules with the needed accuracy to predict energy states and reactivity is hard. IBM performed the numerical simulation on H2, LiH, and BeH2. “While this model of BeH2 can be simulated on a classical computer, IBM’s approach has the potential to scale towards investigating larger molecules that would traditionally be seen to be beyond the scope of classical computational methods, as more powerful quantum systems get built,” noted IBM.
Here’s a good statement of the problem and IBM’s solution taken from the paper (Hardware-efficient variational quantum eigensolver for small molecules and quantum magnets):
“Finding exact solutions to such problems numerically has a computational cost that scales exponentially with the size of the system, and Monte Carlo methods are unsuitable owing to the fermionic sign problem. These limitations of classical computational methods have made solving even few-atom electronic-structure problems interesting for implementation using medium-sized quantum computers. Yet experimental implementations have so far been restricted to molecules involving only hydrogen and helium.
“Here we demonstrate the experimental optimization of Hamiltonian problems with up to six qubits and more than one hundred Pauli terms, determining the ground-state energy for molecules of increasing size, up to BeH2. We achieve this result by using a variational quantum eigenvalue solver (eigensolver) with efficiently prepared trial states that are tailored specifically to the interactions that are available in our quantum processor, combined with a compact encoding of fermionic Hamiltonians and a robust stochastic optimization routine.”
There are, of course many approaches to quantum computing and new kinds of qubits seem to appear weekly. IBM, Microsoft, and Google are focused on so-called universal quantum computers able to do pretty much anything classical computers can do. D-Wave builds quantum annealing computers best suited for certain optimization problems, some of which include quantum chemistry problems.
“IBM, Google and a number of academic labs have chosen relatively mature hardware, such as loops of superconducting wire, to make quantum bits (qubits). These are the building blocks of a quantum computer: they power its speedy calculations thanks to their ability to be in a mixture (or superposition) of ‘on’ and ‘off’ states at the same time.”[i] Microsoft is pursing one of the more exotic approaches – a topological qubit, the Majorana, a particle whose existence has been debated but for which evidence has been rapidly accumulating recently.
As described by IBM’s work, the fundamental goal in electronic-structure problems is to solve for the ground-state energy of many-body interacting fermionic Hamiltonians. Solving this problem on a quantum computer relies on a mapping between fermionic and qubit operators, which restates the problem as a specific instance of a local Hamiltonian problem on a set of qubits.
“Here we introduce and implement a hardware-efficient ansatz preparation for a VQE (variational quantum eigensolvers), whereby trial states are parameterized by quantum gates that are tailored to the physical device that is available. We show numerically the viability of such trial states for small electronic-structure problems and use a superconducting quantum processor to perform optimizations of the molecular energies of H2, LiH and BeH2, and extend its application to a Heisenberg antiferromagnetic model in an external magnetic field,” write the authors, all from IBM Research.[ii]
Below is diagram and caption of the recent work taken from the paper. (inset after)
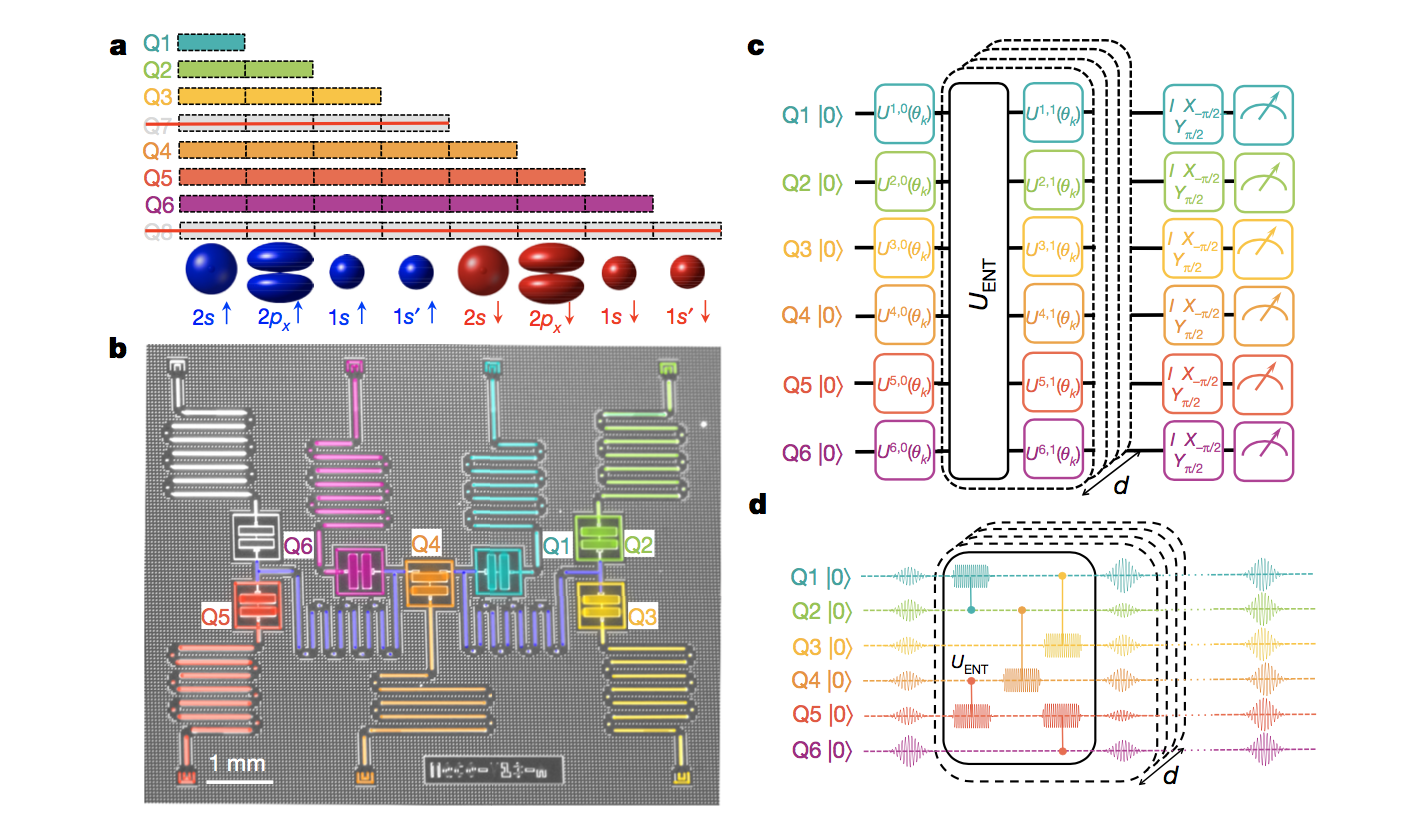
spin and parity symmetries. The length of the bars indicate the parity of the spin orbitals that are encoded in each qubit. b, False-coloured optical micrograph of the superconducting quantum processor with seven transmon qubits. These qubits are coupled via two coplanar waveguide resonators (violet) and have individual coplanar waveguide resonators for control and read-out. c, Hardware-efficient quantum circuit for trial- state preparation and energy estimation, shown here for six qubits. For each iteration k, the circuit is composed of a sequence of interleaved single-qubit rotations Uq,d(θk) and entangling unitary operations UENT that entangle all of the qubits in the circuit. A final set of post-rotations
(I, X−p/2 or Yp/2) before the qubits are read out is used to measure the expectation values of the individual Pauli terms in the Hamiltonian and to estimate the energy of the trial state. d, An example of the pulse sequence for the preparation of a six-qubit trial state, in which UENT is implemented as a sequence of two-qubit cross-resonance gates.
IBM has certainly been an industry leader in providing access to quantum computing, most visibly through its IBM Q initiative launched a year ago with a robust five-qubit quantum computer on the cloud for anyone to freely access; it has recently upgraded to a 16-qubit processor available for beta access.
To help showcase how quantum computers are adept to simulating molecules, developers and users of the IBM Q experience are now able to access a quantum chemistry Jupyter Notebook. The open source quantum chemistry Jupyter Notebook (available through the open access QISKit github repo) allows users to explore a method of ground state energy simulation for small molecules such as hydrogen and lithium hydride.
Quoted in the IBM announcement of the most recent work, Alán Aspuru-Guzik, professor of chemistry and chemical biology at Harvard University characterized IBM’s recent work as impressive, noting “When quantum computers are able to carry out chemical simulations in a numerically exact way, most likely when we have error correction in place and a large number of logical qubits, the field will be disrupted. Exact predictions will result in molecular design that does not need calibration with experiment. This may lead to the discovery of new small-molecule drugs or organic materials.”
Link to IBM paper: http://www.nature.com/nature/journal/v549/n7671/full/nature23879.html?foxtrotcallback=true
[i] http://www.nature.com/news/inside-microsoft-s-quest-for-a-topological-quantum-computer-1.20774
[ii] Abhinav Kandala, Antonio Mezzacapo, Kristan temme, Maika takita, Markus Brink, Jerry M. Chow1 & Jay M. Gambetta


























































