Datanami
Datanami EnterpriseAI
EnterpriseAI HPCwire Japan
HPCwire Japan QCwire
QCwire HPC & AI Wall Street
HPC & AI Wall Street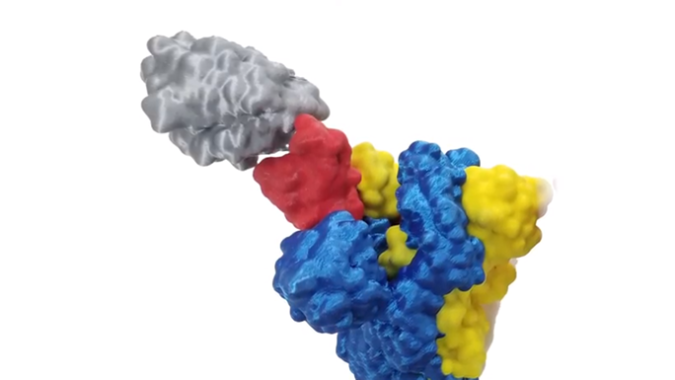
A wide range of supercomputers are crunching the infamous “spike” protein of the novel coronavirus, from Summit more than a month ago to Folding@home to a Russian cluster just a week ago. Now a new player has entered the arena, with a team of researchers from Stony Brook University preparing to bring their spike protein simulations to the big leagues.
COVID-19’s spike protein is both its namesake (the spines around the virus make it look like a crown) and the enabler of its pandemic nature. The spike protein feels around for human cells that it can attach to, after which it transforms and enters the cell, allowing it to replicate. Researchers have therefore been focusing on ways to disable or otherwise interfere with the spike protein in the hopes of producing effective therapeutics or vaccines to combat the disease.
Carlos Simmerling, professor of chemistry and associate director of the Laufer Center at Stony Brook University, is leading another assault on the spike protein. “What we would like to be able to do is to develop small-molecule drugs that can work quickly with the spike and stop it from changing its shape,” Simmerling said. To that end, his team is examining the spike protein in excruciating detail, building on previous work that produced static atomic-level images of the protein and simulated based elements of its behavior.
“You may know that your car door is the way you can get in and out of a car,” Simmerling said, “but if you’ve only seen the door in a picture and have never watched someone actually open a door, you don’t really know how it works. With our research and computer modeling, we are working to understand the process at that more detailed level. For example, what exactly leads to the spike of the virus opening the ‘door’ to infection when it contacts the cell? And, are there places on the spike away from this contact point that could act like keys, where scientists might be able to make a small molecule that would bind to it and lock it closed?”
Now, Simmerling’s team is ready to move their models to more powerful systems. Computational science teams at Argonne National Laboratory and Brookhaven National Laboratory are preparing to run the models on their supercomputers, which include the 8.6 Linpack petaflops Mira, the 6.9 Linpack petaflops Theta and the 17.2 Linpack petaflops Sequoia.
“We will be looking at 60 different target sites where a new drug may attach to the virus,” explained Kerstin Kleese van Dam, director of Brookhaven Lab’s Computational Science Initiative, “and 1 billion drug-like molecules that might be used to modify the ‘door handles,’ ‘hinges,’ or other components so we can identify the most promising options for neutralizing the virus to keep it from entering cells.”
Header image: the spike protein latches onto a cell. Image courtesy of the researchers.
To read Brookhaven National Laboratory’s article discussing this research, click here.