Datanami
Datanami EnterpriseAI
EnterpriseAI HPCwire Japan
HPCwire Japan QCwire
QCwire HPC & AI Wall Street
HPC & AI Wall Street
Most models of SARS-CoV-2, the coronavirus that causes COVID-19, hone in on key features of the virus: for instance, the spike protein. Some of this is attributable to the relative importance of those features, but most is attributable to computational necessity – simulating even a single protein in such excruciating resolution is extraordinarily computation-intensive. Now, researchers from the University of Chicago and the University of California, San Diego have developed the first complete model of the coronavirus, finding, among other things, that its spike proteins move cooperatively.
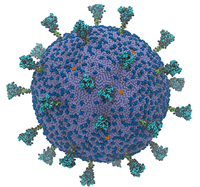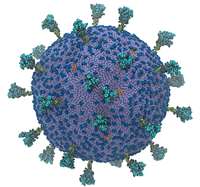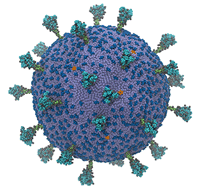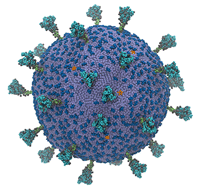
“We wanted to understand how SARS-CoV-2 works holistically as a whole particle,” said Gregory Voth, a professor of chemistry at the University of Chicago and corresponding author of the study, in an interview with Jorge Salazar of the Texas Advanced Computing Center (TACC). “We developed a bottom-up coarse-grained model where we took information from atomistic-level molecular dynamics simulations and from experiments.”
Voth and his colleagues have been using the coarse-grained modeling method, which resolves groups of atoms rather than each individual atom, to study viruses like HIV and the flu for decades. “The benefit of the coarse-grained model is that it can be hundreds to thousands of times more computationally efficient than the all-atom model,” he said. “What you’re left with are the much slower, collective motions. The effects of the higher frequency, all-atom motions are folded into those interactions if you do it well. That’s the idea of systematic coarse-graining.”

Using the coarse-graining method, the team simplified and combined atomistic models of SARS-CoV-2’s spike, membrane, nucleocapsid and envelope proteins. Some of these all-atom models were generated by the Amaro Lab (led by Gordon Bell Special Prize winner Rommie Amaro) using the 23.5 Linpack petaflops Frontera supercomputer at TACC, and the remaining models were generated on the Anton 2 system at the Pittsburgh Supercomputing Center (PSC).
“Frontera and Anton 2 provided the key molecular level input data into this model,” Voth said. “Frontera has been especially useful in providing the molecular dynamics data at the atomistic level for feeding into this model. It’s very valuable.”
The supercomputer-powered coarse-grained model showed some interesting behavior: the virus’ spike proteins, which litter its surface and allow it to infect human cells, move cooperatively.
“They don’t move independently like a bunch of random, uncorrelated motions. They work together,” Voth said. “The paper we published shows the beginnings of how the modes of motion in the spike proteins are correlated. … The ultimate goal of the model would be, as a first step, to study the initial virion attractions and interactions with ACE2 receptors on cells and to understand the origins of that attraction and how those proteins work together to go on to the virus fusion process.”
To learn more about this research, read the paper here and read the article by TACC’s Jorge Salazar here.