Datanami
Datanami EnterpriseAI
EnterpriseAI HPCwire Japan
HPCwire Japan QCwire
QCwire HPC & AI Wall Street
HPC & AI Wall Street
Using the advanced computational resources at the Texas Advanced Computing Center (TACC) at The University of Texas at Austin, researchers uncovered a link between Alzheimer’s disease and cancer that may pave the way for better treatment options and new medicines. The two afflictions share a pathway in gene transcription, a process essential for cell reproduction and growth. The team, led by Houston Methodist Research Institute (HMRI), published its findings in December 2013 in the open access journal Scientific Reports by the Nature Publishing Group.
The scientists used TACC’s Lonestar and Stampede supercomputers to analyze and compare data from thousands of genes, looking for common cell signaling pathways shared by the two diseases. The Lonestar and Stampede systems are part of the Extreme Science and Engineering Discovery Environment (XSEDE), a virtual science environment that supports the interactive sharing of compute resources, data and expertise. Funding for the research comes from the T.T. and W.F. Chao Foundation, and by grants from the National Institutes of Health (NIH).
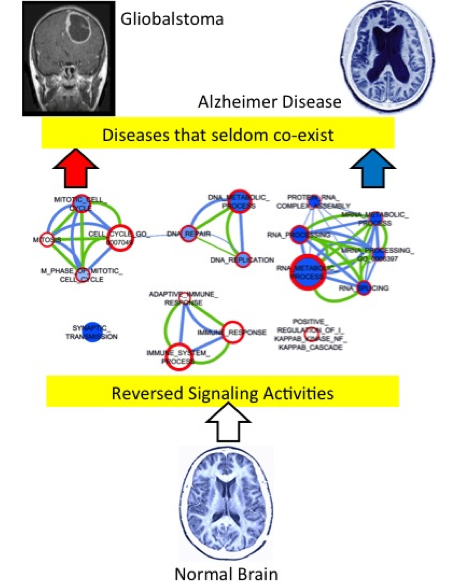
When comparing with normal brain tissues, the microarray profiles for brain tissues from Alzheimer’s disease and GBM patients show significantly reversed signaling activities, highlighted by Gene Ontology terms (nodes) enriched with genes down-regulated in AD (shown as blue in the node face, with darker color indicate larger fold changes) and up-regulated in GBM (red in the node boundary). Credit: Image and caption used with permission by Stephen Wong.
According to lead investigator Stephen Wong, a medical researcher and bioengineer with HMRI, the study is the first to establish a link at the molecular level between Alzheimer’s disease, the most prevalent type of neurodegenerative disease, and glioblastoma multiform (GBM), the most aggressive type of brain cancer.
Earlier studies in 2012 and 2013 found an inverse association between Alzheimer’s disease, which is characterized by nerve cell death and tissue loss in the brain, and with cancer, which occurs when abnormal cells grow and spread very fast. The data pointed to a common genetic pathway, but the details weren’t there.
“No one understands why this link is there, in a biological sense,” Wong said. “And that’s the reason we did this study. I think we are among the first to study it this way.”
The first step in finding the common genes expressed in each disease is to use DNA microarray to reveal the active and inactive genes shared between the two diseases.
The active genes are then mapped to known pathways through a process called pathway analysis. The group began with a working list of potential common pathways and narrowed this down through validation tests performed with cell cultures and live mice.
Knowing this pathway will be a huge step forward in the search for new therapies for this debilitating and deadly diseases.
The results of this study show that the ERK/MAPK cell signal pathway is up-regulated in brain cancer, while the Angiopoietin Signaling pathway is up-regulated in Alzheimer’s disease. In Alzheimer’s cells from mice, tumor suppression is mediated by the ERK-AKT-p21-cell cycle pathway and anti-angiogenesis pathway.
“Although GBM and Alzheimer’s both affect nearly 50% for aged population between 65 and 85 years of age, the body itself has very fine regulation at a very detailed level within the individual signaling pathways to make these two diseases exclude each other,” said study co-author Hong Zhao with the HMRI. “Different kinds of cells, like Alzheimer’s disease cells or cancer cells, have very fine and elaborated regulations on the general molecular signaling pathways, which depend on the cells’ response to the microenvironments.”
The study relied on microarray data covering 524 AD and 1,091 GBM subjects. The analysis included gene annotation, pathway expansion, enrichment analysis, and other details, which was enabled by TACC’s powerful supercomputers.
From this data set, the scientists identified more than 2,000 significant genes with 15 gene ontology terms marked as significantly changed.
“TACC helped us in accomplishing data analysis. We’re using TACC’s Lonestar and Stampede supercomputing clusters to do all this number crunching,” Wong said.
While this study mainly looked at “fairly manageable” data sets of microarray data, the next stage will require that the team analyze much more fine-grained and computationally costly gene sequencing data.
“The gene sequencing data size would easily be 1000-fold larger than the microarray data in the reported study,” Wong said, “which means the need to use TACC’s Lonestar and Stampede supercomputing clusters for number crunching is even more eminent.”